For big data people
Parallelization
The time limiting part are HMMER (search genes). If you want to deal with a large data
a collection of file containing replicons (each file must contains one replicon)
or a gembase file (with a lot of replicons, from ten to more than thousand)
we provide a workflow to parallelize the execution by the data. This mean that
We split the data input into chunks containing one replicon each (for gembase input file).
Then execute MacSyFinder in parallel on each replicon (the number of parallel tasks can be limited)
Then aggregate the results in one global summary.
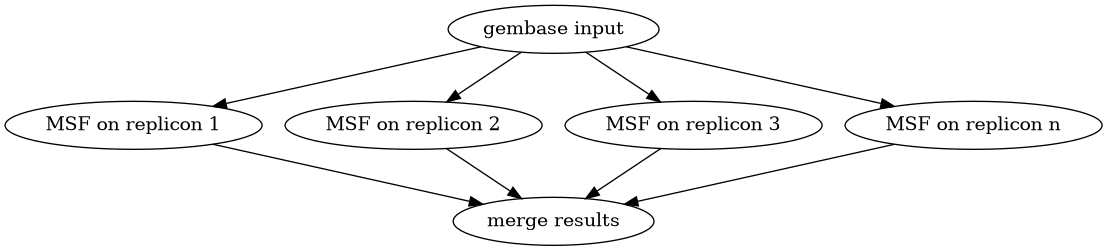
Diagram of the parallel_macsyfinder workflow on gembase input
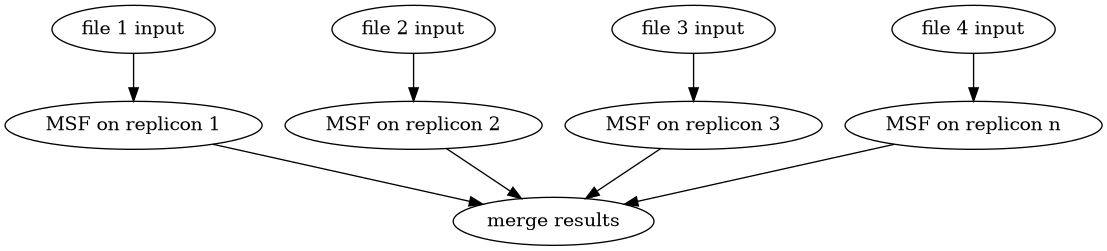
Diagram of the parallel_macsyfinder workflow on ordered or unordered replicon
The workflow use the nextflow framework and can be run on a single machine or a cluster.
First, you have to install nextflow first, and macsyfinder. Then we provide 2 files (you need to download them from the MacSyFinder github repo.)
parallel_macsyfinder.nf which is the workflow itself in nextflow syntax
nextflow.config which is a configuration file to execute the workflow.
The workflow file should not be modified. Whereas the profile must be adapted to the local architecture.
- The file nextflow.config provide five profiles:
a standard profile for local use (single machine).
an apptainer profile using docker image with apptainer executor (on a single machine).
a docker profile using docker image with docker executor (on a single machine).
a cluster profile.
a cluster profile using apptainer container system with the docker image.
How to get parallel_macsyfinder
The release contains the workflow parallel_macsyfinder.nf and the nextflow.config at the top level of the archive But if you use pip to install MacSyFinder you have not easily access to them. But they can be downloaded or executed directly by using nextflow.
to download it
nextflow pull gem-pasteur/macsyfinder
to get the latest version or use -r option to specify a version
nextflow pull -r release_2.0 gem-pasteur/macsyfinder
to see what you download
nextflow view macsyfinder
to execute it directly on a local host with macsyfinder already installed and with with models installed too:
nextflow run gem-pasteur/macsyfinder -profile standard --models "TFF-SF all" --db-type gembase --sequence-db <path/to/my/gembase.fasta>
or:
nextflow run -r release_2.0 gem-pasteur/macsyfinder -profile standard --models "TFF-SF all" --db-type gembas --sequence-db <path/to/my/gembase.fasta>
or for ordered replicon
nextflow run gem-pasteur/macsyfinder -profile cluster_apptainer --models "TFF-SF all" --db-type ordered_replicon --sequence-db '<path/to/replicons/*.fasta>' --outdir <my_results>
nextflow run gem-pasteur/macsyfinder -profile cluster_apptainer --models "TFF-SF all" --db-type ordered_replicon --sequence-db 'file1.fasta,file2.fasta,file3.fst' --outdir <my_results>
or if you download the macsyfinder repository, or the the workflow with it’s configuration file:
nextflow run parallel_macsyfinder.nf -profile standard --models "TFF-SF all" --db-type ordered_replicon --sequence-db 'data/base/split/GCF_*.fasta' --outdir GCF
Note
For gembase data the workflow expected one file with several replicons.
For ordered_replicon or unordered the workflow expected several files with one replicon per file.
Warning
See the double quotes surrounding the models value –models “TFF-SF all” with out quoting macsyfinder will not received the right argument.
Warning
See the (double) quotes surrounding the models value –sequence-db ‘<path/to/replicons/.fasta>’* with out quoting parallel_macsyfinder will not received all files.
Warning
When you analyzed ordered or unordered replicons (–db-type set to ordered_replicon or unordered) the –out-dir option is REQUIRED.
standard profile
This profile is used if you want to parallelize MacSyFinder on your machine. You can specify the number of tasks in parallel by setting the queueSize value You can also fix the number of cpu used by each task (macsyfinder –worker option see macsyfinder options) by setting the params.worker parameter in nextflow.config
standard {
executor {
name = 'local'
queueSize = 4
}
process {
errorStrategy = 'ignore'
withName: macsyfinder {
cpus = params.worker
}
}
}
Almost options available in non parallel version are also available for the parallel one.
except:
* --db-type which is set to gembase (only data type supported for the parallelized macsyfinder version).
* --out-dir which is not available.
A typical command line will be:
./parallel_macsyfinder.nf -profile standard --models "TFF-SF all" --sequence-db <path/to/my/gembase.fasta>
Note
The options starting with one dash are for nextflow workflow engine, whereas the options starting by two dashes are for macsyfinder workflow.
If you execute this line, 2 kinds of directories will be created.
One named work containing lot of subdirectories this for all jobs launch by nextflow.
Directories named merged_macsyfinder_results_XXX where XXX is the name of the gembase file. This directory contain the final results as in non parallel version.
standard_apptainer or standard_docker profile
If you have not installed macsyfinder but you use it through a container docker or https://apptainer.org/ (former singularity) We provide profiles for these situations. With the command line below nextflow will download parallel_macsyfinder from github and download the macsyfinder image from the docker-hub (https://hub.docker.com/r/gempasteur/macsyfinder) (and apptainer convert the image on the right format on the fly) so you haven’t to install anything except nextflow and apptainer or docker.
standard_apptainer {
executor {
name = 'local'
queueSize = 4
}
process {
errorStrategy = 'ignore'
container = 'docker://gempasteur/macsyfinder:latest'
withName: macsyfinder {
cpus = params.worker
}
}
singularity {
enabled = true
}
}
standard_docker {
executor {
name = 'local'
queueSize = 4
}
process {
errorStrategy = 'ignore'
container = 'macsyfinder'
withName: macsyfinder {
cpus = params.worker
}
}
docker {
enabled = true
runOptions = '--user $(id -u):$(id -g)'
}
}
The execution is similar than for installed macsyfinder
./parallel_macsyfinder.nf -profile standard_apptainer --models "TFF-SF all" --sequence-db <path/to/my/gembase.fasta>
or
./parallel_macsyfinder.nf -profile standard_docker --models "TFF-SF all" --sequence-db <path/to/my/gembase.fasta>
cluster profile
The cluster profile is intended to work on a cluster managed by SLURM. If your cluster is managed by an other drm replace executor name by the right value (see nextflow supported cluster )
You can also manage
The number of tasks in parallel with the executor.queueSize parameter (here 500). If you remove this line, the system will send in parallel as many jobs as there are replicons in your data set.
The queue (or partition in Slurm teminology) with process.queue parameter (here common,dedicated)
and some options specific to your cluster management systems with process.clusterOptions parameter
cluster {
executor {
name = 'slurm'
queueSize = 500
}
process {
errorStrategy = 'ignore'
queue = 'common,dedicated'
clusterOptions = '--qos=fast'
withName: macsyfinder {
cpus = params.worker
}
}
}
To run the parallel version on cluster, for instance on a cluster managed by slurm, I can launch the main nextflow process in one slot. The parallelization and the submission on the other slots is made by nextflow itself. Below a command line to run parallel_macsyfinder and use 3 cpus per macsyfinder task, each macsyfinder task can be executed on different machine, each macsyfinder task claim 2 cpus/cores (cpu in nextflow terminology/ cores for hardware) to speed up the genes search.
sbatch --qos fast -p common nextflow run parallel_macsyfinder.nf -profile cluster --models "TFF-SF all" --sequence-db <path/to/my/gembase.fasta> --worker 3
The results will be the same as describe in local execution.
cluster_apptainer profiles
You can also use the macsyfinder apptainer image on a cluster, for this use the profile cluster_apptainer.
sbatch --qos fast -p common nextflow run gem-pasteur/macsyfinder -profile cluster_apttainer --models "TFF-SF all" --sequence-db <path/to/my/gembase.fasta>
In the case of your cluster cannot reach the world wide web. you have to download the singularity image
apptainer pull --name macsyfinder.simg docker://gempasteur/macsyfinder
Then move the image on your cluster modify the nextflow.config to point on the location of the image, and adapt the cluster options (executor, queue, …) to your architecture
cluster_apptainer {
executor {
name = 'slurm'
queueSize = 500
}
process {
errorStrategy = 'ignore'
container = '/path/to/macsyfinder.simg'
queue = 'common,dedicated'
clusterOptions = '--qos=fast'
withName: macsyfinder {
cpus = params.worker
}
}
singularity {
enabled = true
runOptions = '-H $HOME -B /pasteur'
autoMounts = false
}
}
then run it
sbatch --qos fast -p common nextflow run ./parallel_macsyfinder.nf -profile cluster_apptainer --models "TFF-SF all" --sequence-db <path/to/my/gembase.fasta>
If you want to have more details about the jobs execution you can add some options to generate report:
Execution report
To enable the creation of this report add the -with-report command line option when
launching the pipeline execution. For example:
nextflow run ./parallel_macsyfinder.nf -profile standard -with-report [file name] --models "TFF-SF all" --sequence-db <path/to/my/gembase.fasta>
It creates an HTML execution report: a single document which includes many useful metrics about a workflow execution. For further details see https://www.nextflow.io/docs/latest/tracing.html#execution-report
Trace report
In order to create the execution trace file add the -with-trace command line option when launching the pipeline
execution. For example:
nextflow run ./parallel_macsyfinder.nf -profile standard -with-trace --models "TFF-SF all" --sequence-db <path/to/my/gembase.fasta>
It creates an HTML timeline for all processes executed in your pipeline. For further details see https://www.nextflow.io/docs/latest/tracing.html#timeline-report
Timeline report
To enable the creation of the timeline report add the -with-timeline
command line option when launching the pipeline execution. For example:
nextflow run ./parallel_macsyfinder.nf -profile standard -with-timeline [file name] --models "TFF-SF all" --sequence-db <path/to/my/gembase.fasta> ...
It creates an execution tracing file that contains some useful information about each process executed in your pipeline script, including: submission time, start time, completion time, cpu and memory used. For further details see https://www.nextflow.io/docs/latest/tracing.html#trace-report
Warning
When you run parallelize version of macsyfinder the hhm score for each genes can be different than in non parallel version. As hmmsearch use the size of the sequence database to compute the score.
